
期刊:Science China-Life Sciences
影响因子:8.0
在前面的章节中,我们系统地回顾了单细胞组学的最新进展。尽管这些单细胞测序技术允许以前所未有的分辨率调查细胞异质性,但它们远远不足以充分了解多细胞生物的复杂工作原理。许多研究强调,一个细胞的状态不仅受到细胞内调节网络的调节,还受到来自环境的细胞外信号的干扰。在实验过程中,组织的分离和单个细胞的分离都会导致关键空间信息的丢失,包括细胞位置及其相互接近度。空间转录组学(ST)解决了这一限制,使测量基因表达与空间信息保存。在本节中,我们将介绍空间转录组学技术,讨论空间数据分析的计算方法,并回顾其在各种生物系统中的应用。此外,我们还将深入探讨空间多组学技术的最新进展。
空间分辨转录组学技术
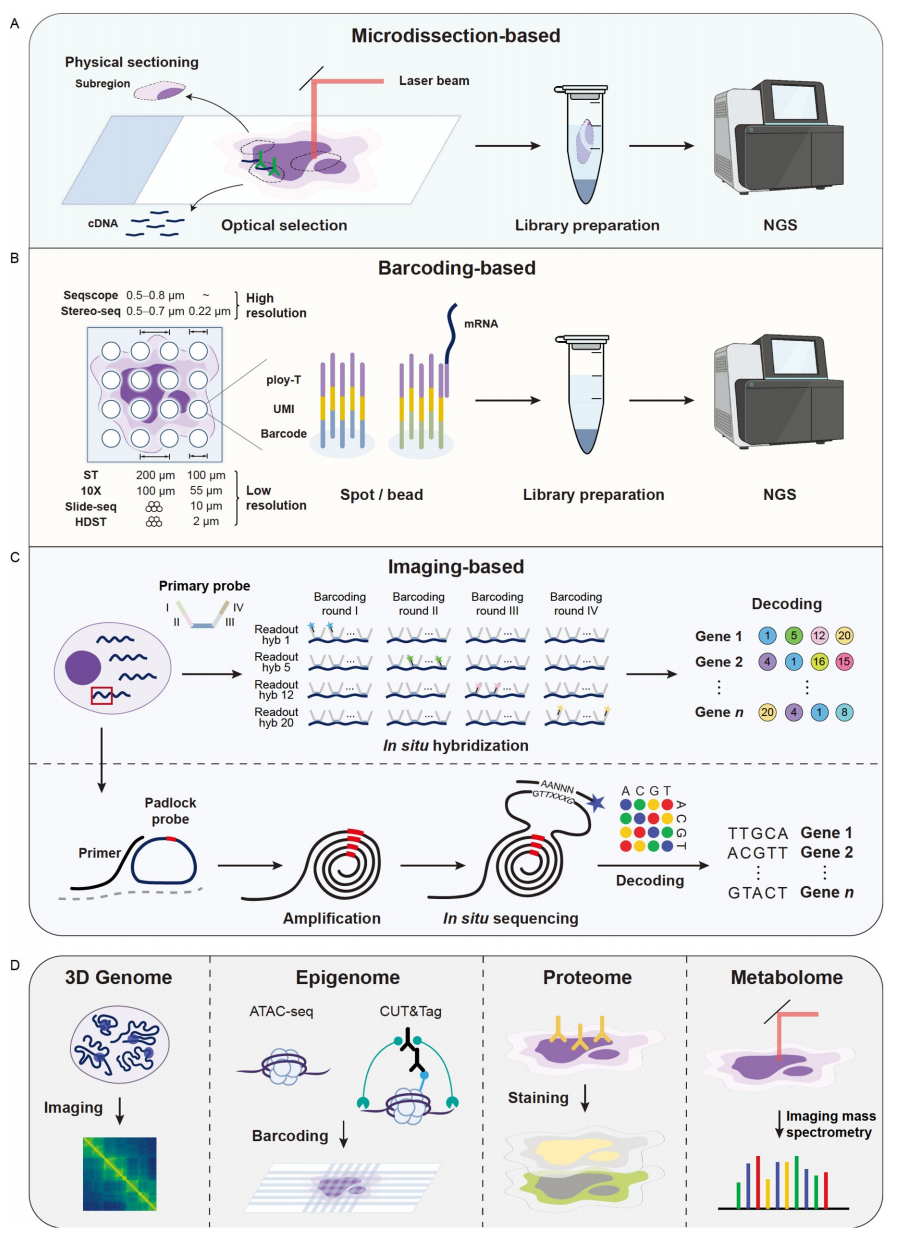
图14
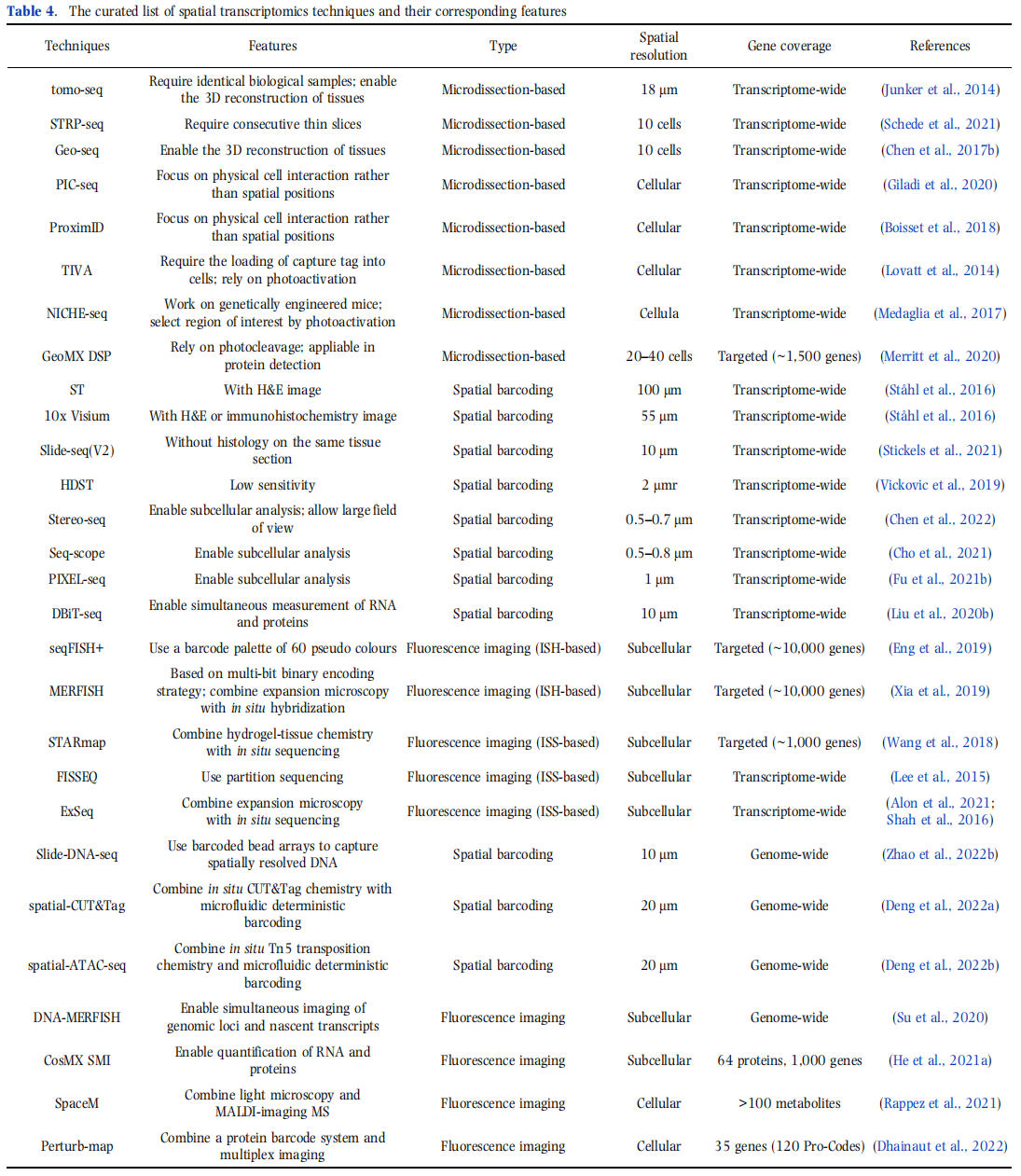
表4
1. 基于显微解剖的ST技术
属于这一类的技术旨在通过各种显微解剖方法从多个空间近端组织亚区中计算重建组织的三维结构(图14A)。例如,RNA断层扫描(tomoseq)从多个假定相同的生物样本中沿着三个正交轴的一系列顺序冷冻中获得RNA。相同生物样本的要求限制了tomo-seq在人类样本上的应用。相比之下,STRP-seq采用两级解剖策略将组织切片分为初级切片和次级切片,假设空间表达模式在间隔14 μm的连续初级切片之间是恒定的。基于冷冻切片,Geo-seq利用LCM将组织切片成小至10个细胞左右的区域。这类方法中的其他方法包括ProximID 和PIC-seq,它们侧重于两个(双胞胎)或三个细胞(三胞胎)内的物理细胞相互作用,而不是组织中的位置或周围环境。
除了物理切片,显微解剖可以通过结合光学标记和基于荧光的细胞选择,或基因指数寡核苷酸的光切割来完成。例如,转录组体内分析(TIVA)加载TIVA标签(即光激活的mRNA捕获分子)进入活细胞,并通过激光光激活选择细胞,随后触发标签与mRNA的杂交。作为一种替代技术,NICHE-seq将标记的地标细胞注射到表达光激活绿色荧光(PA-GFP)的转基因小鼠中,允许对感兴趣的生态位进行原位标记。组织解离后,活化的PA-GFP+细胞通过FACS进行分类,进行单细胞转录组分析。NanoString开发的商用GeoMX数字空间轮廓仪(DSP)采用带有UV可切割接头的探针,并自动进行光学选择。
总的来说,微解剖与单细胞或bulk-RNA相结合测序使得在空间背景下研究转录组成为可能。显微解剖可以用物理方式进行,也可以用光学方式进行。物理切片通常是手工进行的,这使得解剖方案既费力又耗时。相比之下,光学依赖切片通常依赖于将专门的标签加载到活细胞或模式生物的基因工程中,这限制了其在新鲜冷冻或FFPE人类样本中的应用。无论如何进行显微解剖和测序,在所选择的子区域内的剖面细胞的确切位置是未知的,导致普遍较低的空间分辨率。
2. 基于条形码的ST技术
基于显微解剖的技术通过手动标记每个子区域来跟踪空间信息。空间条形码技术可以自动记录空间坐标(图14B)。在这种方法中,条形码与UMIs和聚寡核苷酸一起固定在玻璃载玻片上,以便原位捕获mRNA和cDNA合成。阵列中的每个条形码点直径为100 μm,距离相邻点的中心距离为200 μm,分辨率为10-40个单元。10x Genomics利用直径为55 μm、中心到中心距离为100 μm的斑点进一步将空间分辨率提高到5-10个细胞。一些技术不是将条形码直接附着在载玻片上,而是将条形码与小珠连接起来,用于位置标记和mRNA捕获。例如,Slide-seq将10 μm的dna条形码珠沉积在表面上。同样,HDST将条形码珠放入2 μm井的阵列中。这两种技术都将空间分辨率提高到1-2个单元。然而,由于条形码珠粒是随机分布在载玻片上的,因此需要原位测序(ISS)或原位杂交(ISH)来解码每个固定珠粒的条形码序列。尽管基于头部的技术可以达到细胞分辨率,但它们仍然过于粗糙,无法检测亚细胞差异。
最近,通过重新利用Illumina测序平台,开发了Seq-scope,以实现0.5-0.8 μm的中心到中心分辨率。另一种实现亚微米分辨率分析的技术是Stereo-seq,其中包含条形码的220纳米DNA纳米球(dnb)沉积在中心距离为500或715纳米的图案阵列上。Seq-scope和Stereo-seq都需要两轮测序,其中第一轮将条形码与空间位置相关联,第二轮提供捕获cDNA的信息,就像Slide-seq一样。
总之,基于条形码的方法将空间条形码技术与NGS相结合,允许在空间背景下对RNA进行转录组分析。这些技术涉及空间分辨率和检测效率之间的权衡。与最初的ST技术或商业化的10倍Visium相比,Seq-scope、Stereo-seq在空间分辨率上的提高往往是以低检测灵敏度和低基因覆盖率为代价的。
3. 基于成像的ST技术
基于微解剖和基于条形码的技术在位置标记后提取核酸分子用于NGS测序。为了在原位保存RNA,各种原位转录组学技术被开发出来用于基因表达的空间定位,包括ISH和ISS(图14C)。由于这些方法需要荧光成像,因此它们被统称为基于成像的技术。大多数基于ish的ST技术主要依靠单分子RNA荧光原位杂交(smFISH)来实现靶向转录物的原位定量测量。SeqFISH属于这种类型,它可以通过连续的荧光杂交、成像和剥离读出探针来同时检测多个mRNA分子。使用seqFISH策略,所有的目标基因都是通过几轮读出探针的组合来编码的。SeqFISH+将读出探针调色板从SeqFISH中的四种或五种颜色扩展到60种“伪颜色”,从而在单个细胞中实现多达10000个基因的多路复用。MERFISH是另一种基于smfish的技术,它也需要多轮杂交,但采用了独特的多位二进制编码策略。为了解决光学拥挤问题,将扩展显微镜(ExM)集成到MERFISH中。编码策略,结合ExM,允许MERFISH减少杂交轮数。例如,为了保证检测到10,000个基因,使用三色成像,seqFISH+需要80轮(4×20)杂交,而MERFISH只需要23轮就能构建一个汉明权值为4的69位HD4编码。除了基于多路FISH的技术外,ISS也可以实现RNA的原位分析,它通过原位信号扩增对固定组织或细胞样本中的RNA进行测序。由于细胞空间有限,一些基于isss的技术通过设计针对特定RNA或cDNA的探针来选择部分基因。2013年发表的最初的ISS方法使用挂锁探针与靶标结合,然后通过滚环扩增(RCA)产生RCA产物,用于后续的结扎测序。STARmap使用双组分挂锁探针直接结合RNA而不是cDNA,避免了RNA到cDNA的低效步骤,降低了潜在的噪声。为了消除传统的支持寡核苷酸连接检测(SOLiD)测序带来的强背景荧光,STARmap设计了动态退火和连接减错测序(SEDAL),可以在测序过程中抑制误差。
除了靶向ISS方法外,还可以采用非靶向方式进行ISS,即将转录物反向转录为cDNA,然后进行DNA扩增和测序,而不需要对基因进行预选择。虽然非靶向方式可以提高转录组的覆盖范围,但它也可能导致分子拥挤。为了缓解这一问题,FISSEQ利用了分区测序策略,其中只有一小部分扩增子被随机选择并使用扩展测序引物进行测序,因此导致检测效率较低。结合ExM, FISSEQ适用于另一种称为ExSeq的方法,以区分拥挤的分子并提高空间分辨率。
一般来说,基于成像的技术提供高空间分辨率,达到细胞甚至亚细胞水平。在这些技术中,基于ish的技术依赖于目标基因的先验知识,具有较高的检测效率。相比之下,由于国际空间站的局限性,基于国际空间站的技术已经效率相对较低,特别是在没有目标的情况下。此外,大多数这些技术都需要专门的高分辨率成像设备,这可能限制了它们更广泛的适用性。
4. 空间多组学技术
为了对细胞进行更全面的表征,在空间背景下对其他模式的测量已经付出了相当大的努力,包括基因组、表观基因组、蛋白质组、代谢组等(图14D)。ST技术中使用的定位策略已经适应于实现其他组学的空间分析。例如,SlideDNA-seq使用条形码头阵列捕获空间分辨的基因组序列,该阵列最初是为空间RNA分析而开发的。同样,通过将DbiT-seq的微流体确定性条形码策略与原位CUT&Tag化学和Tn5转位化学相结合,开发了spatial-CUT&Tag 和spatial-ATAC-seq 来分析组蛋白修饰和染色质可及性。为了了解其原生环境下的三维染色质构象,设计了一种基于merfish的方法来可视化超过1000个基因组位点,用于高分辨率染色质追踪。
在蛋白质组学领域,蛋白质表达可以很容易地通过多重免疫组织化学(IHC)可视化。免疫组化可以进一步与成像质细胞术或多路离子束成像(MIBI)相结合,允许同时成像约100种蛋白质。此外,感兴趣的蛋白质可以被dna条形码抗体靶向,从而通过NGS进行量化,如GeoMx DSP 。细胞表面蛋白可以被抗体结合而不产生细胞裂解,从而防止RNA受到损伤。因此,无论是单细胞组学还是空间组学,蛋白质组学都可以与转录组学相结合。例如,增强版的10x Visium在mRNA捕获之前进行免疫组化,以实现蛋白质和RNA的共同检测,尽管只允许检测1-2种蛋白质。通过在流动条形码之前将抗体衍生标签添加到固定组织载片,DbiT-seq可以测量mRNA和数十种蛋白质。此外,NanoString还提供CosMx SMI平台,可通过高plex成像对1000种RNA和64种蛋白质进行定量分析。从样品中收集的代谢物通常使用质谱法进行定量。为了研究空间分辨代谢组,基于成像质谱法(IMS)的各种技术已经发展起来。这些技术在从样品分子中产生离子的方式上有所不同,包括MALDI 、DESI和SIMS。例如,SpaceM是一种基于maldi的原位单细胞代谢组学方法。它通过将maldi成像与光学显微镜相结合,然后使用计算方法进行图像分割和配准,解决了将代谢物强度分配给单个细胞的挑战。
除了内在遗传外,许多基因功能还受到空间环境的影响。为了研究空间功能基因组学,Dhainaut建立了一种名为Perturb-map的方法,该方法可以在组织背景下以单细胞分辨率汇集CRISPR筛选。这是通过采用蛋白质条形码系统和多路成像来实现的。
空间转录组学计算方法
图15
1. 去噪增强空间转录组学中的信号
如上所述,许多ST技术面临着与低检测效率和显著噪声相关的挑战。这些问题源于对每个空间单元(即点或头)的浅层测序或保存组织结构所需的复杂实验步骤,或两者的结合。Wang等人在10倍的Visium和Slide-seq数据中表明,信号噪声反映在基因表达的dropouts和膨胀中。虽然已经为scRNA-seq数据开发了去噪方法来解决drop-out问题,但它们往往难以纠正“假的”高表达。此外,这些单细胞方法仅依赖于转录组学数据,因此不能直接应用于整合额外的空间信息。
专门开发了几种计算方法来处理去噪的ST数据。例如,spprod可以通过基于潜在图学习的基于条形码的技术在噪声ST数据中推算基因表达。spprod中的去噪过程包括两个步骤。首先,spprod通过利用空间接近性和表达相似性来构建一个图。重要的是,如果可以的话,从相应的病理图像中提取的特征可以用于图的构建。接下来,spprod通过借用图中显示的邻域的表达信息来纠正每个点/头的基因表达。另一种方法,spARC,采用了类似的基于图形的框架,但证明了其在基于成像的ST技术上的适用性。SiGra也是一种基于图的方法,但采用了不同的方法来构建图。它利用成像、转录组和混合三个图形转换器自编码器以及注意机制,使SiGra能够用多模态空间信息增强稀疏和嘈杂的转录组数据。stLearn中的SME方法还允许整合图像特征来规范化空间基因表达。它采用了一种简单的加权平均策略,根据接近点之间的形态相似性计算权重。Ni等人认为,损失和膨胀不是随机的辍学或膨胀,而是由附近点之间和点之间的mRNA流血造成的,这被称为点交换。为了调整现货交换的影响,他们提出了一种称为SpotClean的方法。SpotClean采用概率框架来模拟给定位点上的基因特异性表达,该框架考虑了该位点组织中存在的reads,并将其读取到其他位点上的出血,同时也将其他位点上的出血去除。作者证明SpotClean可以在ST和10x Visium等技术中准确估计基因特异性UMI计数,其中背景位置可以通过ST幻灯片与匹配的H&E图像之间的比对来识别。
2. 基于成像的ST数据的亚细胞分析
基于成像的ST技术为细胞甚至亚细胞分析提供了很好的机会,但也带来了很大的挑战。对于这些技术,每个测量的像素只代表一个转录本,这不足以推断它属于细胞类型。如何将这些单个像素合并形成细胞或亚细胞结构将具有重要意义。在目前的研究中,有两种主要策略用于高分辨率ST数据的分析:基于分割的方法或无分割的方法。细胞分割最初是在显微镜免疫组化图像处理中提出的,它提供了更多关于细胞数量和细胞形态的信息。这里的细胞分割是基于转录本的稀疏测量来确定细胞边界,即将转录本分配给细胞。传统的细胞分割依赖于从染色图像中提取的特征,包括强度和纹理,其中一些可以代表细胞边界。但是对于RNA的荧光图像,揭示细胞边界需要对细胞膜进行特定的染色,这阻碍了细胞的分割。大多数组选择进行额外的细胞核染色(例如DAPI)来识别假定的细胞核,然后用于指导转录本分配。考虑到基因在细胞核区域的表达可能不等于在整个细胞内的表达,一些组合并,辅助poly(A)染色以告知细胞的体细胞。已经开发了几种计算方法来提供替代解决方案。
例如,Qian等人开发了pciSeq,它利用概率框架将RNA点分配给其原始细胞。具体而言,pciSeq将DAPI图像中的细胞核分割作为细胞的初始近似,并分别通过负二项分布和泊松过程模拟细胞RNA计数和基因-细胞距离。该方法以配对的scRNA-seq为参考,利用变分贝叶斯推理估计转录本同时属于细胞和细胞类型的概率。JSTA是另一种依赖于DAPI染色的初始细胞核分割和匹配的scRNA-seq参考的方法。JSTA还可以利用深度神经网络(deep neural network, DNN)作为分类器,通过迭代像素分配实现联合细胞分割和细胞类型标注。
细胞分割可以以不依赖于scrna的方式实现。例如,Baysor可以仅根据转录本的表达进行细胞分割,同时也支持与scRNA-seq获得的细胞类型特异性表达谱的先验信息整合,以及从共染色图像中分割以改进分割。值得注意的是,Baysor使用马尔可夫随机场(MRF)来限制空间近端分子之间的关系。每个细胞都以高斯分布建模,整个数据集可以看作是细胞特异性分布的混合物,可以通过贝叶斯混合模型(Bayesian mixture models, bmm)进行分离。类似地,Sparcle利用Dirichlet过程混合模型进行初始细胞类型识别,并通过借用相邻像素的信息,迭代地将每个转录本分配给细胞。另一种方法ClusterMap也利用邻域的表达来计算邻域基因组成,然后将细胞分割作为一个点模式分析问题,通过密度峰值聚类(DPC)算法求解。
在细胞分割后,可以像scRNA-seq一样进行细胞水平分析,如差异表达分析和细胞-细胞相互作用。更重要的是,进一步探索细胞内的亚细胞结构成为可能。例如,在细胞分割的基础上,ClusterMap可以使用K-means聚类进一步将细胞分割成包括细胞核和细胞质在内的亚细胞结构。Bento是一个用于ST数据亚细胞分析的工具包,它可以进一步识别RNA转录物的5类亚细胞定位,包括核、细胞质、核边缘、细胞边缘,以及以上都不是。
以上讨论的细胞分割方法便于对基于成像的ST数据进行单细胞分析。然而,挑战来自技术噪声,如不均匀的强度信号和生物变异,包括不同的细胞大小和形状以及不同的细胞密度。这些因素可能在实现准确的细胞分割方面造成困难,可能导致下游分析的偏差。因此,已经开发了几种无分割方法,以便在不执行显式分割的情况下进行稳健分析。大多数方法的目的是将每个分子像素分配给特定的细胞类型,而不是单个细胞。为了实现像素的细胞类型分配,Baysor的作者还提供了一种无分割的方法。它是基于相邻RNA分子可能来自同一细胞的假设,共同反映了相应细胞类型的转录组学特征。他们为每个转录本计算一个邻域组合向量(NCV),通过利用邻域信息有效地增强一个像素的信号。ncv随后被视为“伪细胞”,用于下游聚类和注释分析。SSAM提供了一个类似的解决方案,它通过借用其邻域的信息来估计每个像素的mRNA信号。不同的是,它们应用高斯核的核密度估计(KDE),这与Baysor不同,后者为考虑的最近邻域提供相同的权重。
3. 整合scRNAseq解读细胞类型的空间分布
无论组织样本是单细胞还是空间转录组学,细胞类型注释对于破译细胞组成都是非常必要的。为scRNA-seq设计的注释策略,包括基于表达标记基因的无监督聚类和细胞类型推断,似乎适用于ST数据的分析。不幸的是,由于当前ST技术的限制,这种尝试通常不会奏效。首先,对于基于成像的靶向ST技术,限制性的基因选择和读出噪声的存在会阻碍未知细胞类型的鉴定。其次,对于基于条形码的低分辨率ST数据,每个点的多个细胞或细胞类型混合的测量可能是平均的,这可能会模糊细胞的异质性。最后,对于基于条形码的高分辨率ST数据,低检测效率也对明显聚类和适当的细胞类型标注提出了挑战。因此,在大多数情况下,整合ST数据与匹配的scRNA-seq对于了解细胞类型分布是必要的。通常,积分可以通过两种方法完成:映射或反卷积。细胞定位包括两个方面:将预定义的细胞类型映射到空间位置和将scRNA-seq数据中的细胞映射到组织中。前者将细胞类型标记从scRNA-seq转移到空间转录组学,后者预测来自scRNA-seq的细胞的空间位置,在某些情况下也被视为scRNA-seq的空间重构。对于细胞类型定位,可以使用来自scRNA-seq的细胞类型特异性基因特征来计算富集分数。该方法已被证明在分析基于微阵列的胰腺导管腺癌的ST数据方面是有效的。对于基因有限的基于成像的ST方法,上述的细胞分割方法,如pciSeq 、JSTA 和Baysor 也可以在scRNA-seq可用的情况下进行细胞类型分配。另外,由于这些基于成像的ST技术可以在细胞分割后提供单细胞水平的表达,因此现有的单细胞数据整合方法可以直接应用于单细胞分辨率空间数据和scRNA-seq的整合。例如,Seurat通过典型相关分析(CCA)将细胞从ST和scRNA-seq投射到共享潜在空间。以互近邻(MNN)识别的细胞对为锚点,scRNA-seq的细胞类型标记可以转移到空间细胞中。LIGER和Harmony也可以实现类似的集成。这些单细胞整合方法利用共同的潜在空间和邻域信息,还可以预测ST缺失基因的空间表达,增强ST谱基因原有的弱信号。scRNA-seq的空间重建是在ST技术繁荣之前提出的,该技术通过一些空间地标性基因的表达来预测细胞的空间位置。早期的方法,如Seurat (v1.0),将含有数十个基因的ISH参考数据建模为二值化表达图,然后通过将scRNA-seq衍生的双峰混合模型与二值化表达参考相关联,概率推断出单个细胞的位置。Achim和DistMap也使用二值化的ISH参考,但采用不同的方法来计算细胞位置对应关系。Achim设计了一个评分方案,根据给定细胞中的基因特异性比率来评估细胞与每个空间位置之间的对应关系。
DistMap利用二值化的单细胞基因表达和空间参考计算马修相关系数(Matthew correlation coefficient,MCC)得分,然后将细胞软分配到空间位置。Tangram是最近开发的一种方法,除了基于ish的数据外,它还能够将scRNA-seq与各种技术测量的空间转录组学相匹配。通过最大化scRNA-seq和ST共享的基因表达的相关性,Tangram可以得到一个概率映射矩阵,该矩阵表示在每个空间位置找到每个单个细胞的概率。最近的方法不是对细胞位置对应进行评分,而是将scRNAseq的空间重构问题转化为监督学习问题或优化问题。例如,DEEPsc通过训练基于神经网络的分类器,将空间参考视为scRNA-seq,将细胞映射到空间位置的问题制定为监督分类问题。经过充分训练的DEEPsc网络将来自细胞的特征向量作为输入,并根据来自不同空间位置的似然度预测细胞的空间起源。另一种方法,glmSMA将单元映射框架为凸优化问题。首先,采用拉普拉斯矩阵表示位置到位置的物理距离和细胞到细胞的表达距离。通过最小化每个细胞和相应位置的表达差异,glmSMA最终可以找到从scRNA-seq中的细胞到st中的空间位置的映射。SpaOTsc将细胞映射定义为一个最优运输问题,旨在将细胞到位置的运输成本最小化。SpaOTsc中的运输成本主要基于scRNA-seq和空间参考之间的基因表达差异来衡量,并结合两个惩罚项来处理两个数据集的不平衡样本量,并分别保留每个数据集内的结构。同样,novoSpaRc采用最优转运框架,其核心假设是物理近端细胞具有相似的表达谱。novoSpaRc通过位置到位置的物理距离和细胞到细胞的表达距离的组合来测量运输成本,两者都计算为各自kNN图中的最短路径。通过最小化运输成本,novoSpaRc最终得到了一种映射,通过这种映射,细胞被映射到尽可能保留原始细胞-细胞对应关系的位置,考虑到上述假设。值得注意的是,novoSpaRc还允许在没有参考ST数据时从头重建scRNA-seq。大多数重建方法都是基于物理接近性可以通过表达相似性来反映的假设。然而,该假设不能代表所有细胞的空间分布模式,这使得推断的细胞位置值得怀疑。
细胞类型反褶积旨在估计每个空间位置(即点或头)的确切细胞类型比例,通常用于scRNA-seq和低分辨率条形码的ST数据(如10x Visium)的整合。对于基于高分辨率条形码的ST技术,如Stereo-seq,原始像素级表达式以基于bin的方式聚合,然后将每个bin作为一个新的空间单元进行反卷积分析。目前的ST反卷积方法基本上可以分为四类:回归、因式分解、概率建模和基于图的方法。回归是一种最流行的方法开发的大量rna序列反褶积。由于每个点覆盖的细胞数量有限,在ST数据上直接应用大量RNA-seq反卷积方法会导致来自不相关细胞类型的噪声。为了克服这一问题,基于阻尼加权最小二乘(DWLS)回归的ST反卷积方法spatialDWLS采用了两种措施。首先,在精确估计细胞类型比例之前进行细胞类型富集分析,以确定每个点可能的细胞类型。其次,在对富集的细胞类型进行第一轮反褶积后,去除预测比例较低的细胞类型,进行另一轮反褶积。基于回归的方法高度依赖于每种细胞类型的标记基因的选择。与对细胞类型特异性表达谱进行回归不同,一些方法提出对潜在主题谱进行回归,该主题谱可以通过矩阵分解从单细胞表达数据中分解出来。例如,NMFreg最初是为Slide-seq的细胞类型注释而开发的,它结合了非负矩阵分解(NMF)和非负最小二乘(NNLS)。它使用NMF从预标记的scRNA-seq中获得一个基本的基因因子谱,然后使用NNLS回归计算每个头的因子负荷。由于每个因子都与细胞类型相关联,因此因子负载充当细胞类型比例。SPOTlight采用了类似的策略,但使用了种子NMF,其中使用了细胞类型特异性标记基因和高可变基因(HVG)的组合,并通过scRNA-seq衍生的细胞-细胞类型归属初始化因子-细胞图谱。反褶积也可以通过分解来实现。例如,STRIDE采用主题建模方法LDA,从scRNA-seq中获得细胞类型相关的主题概况。然后,使用预训练的主题模型可以推断每个点的细胞类型组成。Stdeconvolve也基于LDA,但提供了一种无参考的解决方案。CARD建立在NMF的基础上,但通过条件自回归(CAR)模型考虑了点之间的空间相关性,这使得CARD成为一种“空间”反卷积方法。
除了直观的回归或基于因子分解的方法外,概率建模方法已经开发出来,假设细胞或点中的基因表达遵循特定的概率模型。例如,RCTD通过泊松分布对每个位置的基因表达进行建模,并将每个点拟合为单个细胞类型的线性组合。值得注意的是,RCTD还考虑了特定于平台的效果。Cell2location遵循类似的概念,但使用NB分布来模拟基因表达。同样,Stereoscope使用NB模型,但它适用于完整的基因集,而不是一组选定的标记基因。DestVI还使用NB分布来模拟每个基因在细胞或点中的表达,并使用神经网络编码和解码参数。最重要的是,DestVI不仅可以估计细胞类型比例,还可以恢复每个点的细胞类型特异性表达,从而捕获同一类型细胞内的连续表达变化。
除了DestVI之外,还有其他几种基于神经网络的方法。DSTG首先通过随机混合scRNA-seq的细胞生成伪st数据,然后从伪st和实st构建跨点的链接图。利用捕获点之间内在拓扑相似性的链接图,利用半监督图卷积网络(GCN)估计real-ST中每个点内的细胞类型比例。CellDART也生成伪st数据——一种虚拟的细胞混合物——但采用对抗性域适应的思想。CellDART集成了两个基于神经网络的分类器,其中训练源分类器来预测细胞类型组成,训练域分类器来区分真实斑点和伪斑点。通过在训练过程中迭代更新两个分类器,训练良好的CellDART模型可以从真实ST数据中准确估计每个点的细胞类型比例。另一种基于神经网络的方法GraphST采用了不同的策略。GraphST利用一个图对比自监督框架,通过整合空间位置信息和本地上下文来重建ST数据的基因表达。使用自编码器,GraphST可以单独学习scRNA-seq的潜在表示。基于学习到的特征,通过对比学习机制训练出细胞到点的映射概率矩阵,并结合scRNA-seq的细胞类型注释提供对点的细胞类型组成。
4. 空间域识别
除了离散分布,我们还对细胞类型如何在空间上组织形成组织结构和执行功能感兴趣。直观地说,物理上近端的细胞,无论来自相同或不同的细胞类型,都可以构成一个空间结构,通常称为空间域。空间域的识别将有助于我们理解区域内细胞之间的交流及其生物功能。从某种意义上说,空间域可以看作是具有特定空间模式的细胞群。scRNA-seq的标准Louvain聚类方法是基于基因表达相似性构建的图,没有考虑空间信息,在这里不直接适用。一些空间聚类方法改进了基于图的聚类算法,以纳入空间信息。例如,stLearn利用Louvain或K-means进行全局聚类,并通过考虑物理距离执行局部聚类来寻找空间分离的子聚类或合并空间近端单点。另一种方法是MULTILAYER,它在基因模式共表达图上应用Louvain聚类。首先,MULTILAYER通过迭代凝聚策略检测过表达基因的表达模式。基因表达模式在这里被定义为基因在多个连续位置过表达的区域。然后,MULTILAYER构建一个图,其中节点表示先前检测到的基因模式,边缘表示基因模式之间的相似性(即基因共表达程度)。最后,利用Louvain算法将基因共表达模式划分为多个组织群落。
许多空间聚类方法不是以间接方式合并空间信息,而是将空间接近信息编码在MRF中,其中空间依赖关系由Potts模型表示。Zhu等人开发了smfishHmrf,该算法将隐马尔可夫随机场(HMRF)应用于seqFISH数据的空间域识别。他们首先构建一个邻域图来表示细胞之间的空间关系,其中马尔可夫属性只保留直接相邻节点之间的关系。然后,他们通过联合概率分布来建模每个细胞的区域状态,该分布考虑了细胞的基因表达和邻近细胞的区域状态。通过使用期望最大化(EM)求解场平衡参数,smfishHmrf能够检测具有空间相干基因表达的空间域。BayesSpace采用带MRF的全贝叶斯统计模型,以确保同一簇中的点在物理上更接近。BayesSpace通过使用Markov chain Monte Carlo (MCMC)和跨不同簇的固定精度矩阵,能够稳定地估计模型参数,识别空间簇,甚至提高空间转录组学的分辨率。考虑到MCMC是计算密集型的,固定的平滑参数可能会限制不同ST数据集的性能,Yang等人提出了SC-MEB,以实现高效的计算和可调的平滑参数。特别地,他们采用了一种高效的基于迭代条件模型的期望最大化(ICM-EM)方案来估计参数,并通过改进的贝叶斯信息准则(MBIC)来选择聚类数。上述基于磁共振成像的方法都假定隐藏的细胞状态是离散的,这限制了我们对细胞间空间依赖性的理解。相比之下,SPICEMIX将NMF整合到HMRF中,其中观察到的基因表达被建模为潜在因素的线性混合,潜在因素的混合权重被视为隐藏细胞状态。SPICEMIX从另一个角度来理解,它提供了一种考虑空间信息的ST数据降维方法,可以作为下游聚类的基础。基于推断的细胞状态,SPICEMIX进一步应用分层聚类来定义分类细胞类型。
在ST数据分析中,细胞类型聚类和空间域识别可以被视为两个独立的任务。我们上面讨论的大多数方法都专注于识别空间域,除了SPICEMIX,其中空间聚类旨在推断细胞类型,而不与scRNA-seq整合。另一种方法,FICT旨在通过空间聚类推断基于fish的空间转录组学中的细胞类型。具体来说,FICT通过细胞类型特异性的高斯分布来模拟细胞的表达,并通过多项分布来模拟细胞与其相邻细胞之间的关系。FICT能够通过最大化联合概率似然来分配单元簇。同样,BASS也通过细胞类型特异性正态分布来模拟细胞中的基因表达,但同时,它通过特定域的分类分布来模拟细胞类型归属。有了这样一个层次概率框架,BASS可以同时实现细胞类型聚类和空间域检测。
空间转录组学可以看作是一个点图,适合用于基于图的神经网络。许多基于gnn的方法已经被开发出来,通过整合基因表达和空间信息来学习空间转录组学的低维潜在表征,这可以促进下游分析,如空间域识别和空间变量基因的检测。例如,SpaGCN应用GCN来整合多个信息源,包括基因表达、空间位置和组织学。首先,构建一个图来表示点之间的关系,其中节点表示点,通过将组织学图像特征转换到第三个“z”坐标,并将其与点的原始空间坐标(x, y)结合,计算边缘的距离。然后利用卷积层对图中相邻点的基因表达进行聚合。基于聚合的基因表达,实现无监督迭代聚类算法来识别聚类(即空间域)。
其他方法在基础GCN中引入了额外的机制。正如我们在细胞型反卷积一节中讨论的那样,GraphST通过将基因表达与空间位置信息和本地上下文信息相结合,应用图对比自监督框架来学习ST数据的空间潜在表示。另一种方法是SpaceFlow,它将深度图信息集(DGI)框架集成到GCN编码器中。除了基于空间转录组学构建的空间表达图(SEG)外,SpaceFlow还通过随机排列表达构建了表达排列图(EPG)。这两个图都被送入图卷积编码器得到低维嵌入,DGI使编码器通过判别器损失来区分SEG和EPG的嵌入。有些方法采用自编码器进行空间嵌入。例如,SEDR使用深度自编码器网络来学习基因表达的低维潜在表示,然后使用变分图自编码器(VGAE)将其与空间信息集成。STAGATE为自编码器引入了一种注意机制,使边缘权重的自适应学习成为可能。例如,点相似度。stMVC构建了更全面的学习框架。具体来说,stMVC首先通过数据增强和对比学习,从组织学图像中学习视觉特征。然后利用半监督图注意自编码器(SGATE)基于提取的视觉特征和空间基因表达独立学习特定于视图的表示,并通过注意机制整合两个图。stMVC提出的基于注意力的多视图协同学习模型最终学习出一种更加鲁棒的ST数据表示。由于ST数据的空间信号本质,一些方法将空间域识别问题转化为经典的图像分割问题。RESEPT使用GNN从点-点图中学习三维嵌入,将基因表达作为节点的属性,并通过边缘连通性揭示物理邻接性。将每个点的三维嵌入转换为RGB尺度,使得之前为语义分割而设计的CNN可以直接应用于段空间域。另一种方法,Vesalius采用了类似的RGB嵌入策略,但通过UMAP而不是神经网络进行降维。
5. 空间变异基因和基因表达模式的检测
在scRNA-seq分析中,HVG在降维和随后的细胞聚类中起着关键作用。在空间转录组学中,空间可变基因(SVG)的鉴定对于表征复杂组织中的功能组织也很重要。识别SVG就是寻找在空间上表现出很大变异的基因。scRNA-seq中的HVG检测只考虑了高方差而忽略了空间信息,不能直接应用于SVG识别。已经提出了各种计算方法来从空间转录组学中检测SVG。一些方法基于分割的空间域来识别SVG。例如,像我们上面讨论的那样,SpaGCN首先通过集成多个信息源来识别空间域,然后为每个识别的域定义相邻域。空间可变基因是通过使用Wilcoxon秩和检验识别每个目标域和相应相邻域之间的差异表达基因来确定的。大多数方法不依赖于空间域识别,而是直接将空间信息纳入到模型中来研究基因表达的空间变异。根据核心模型,方法大致可分为三类:基于统计建模的方法、基于图的方法和基于其他原理的方法。
1)基于统计建模
Trendsceek将空间表达式建模为标记点过程,其中空间位置被视为二维点过程,位置表达式被视为标记。对于给定的基因和指定的距离,对距离上的所有点对计算点的空间分布与其标记之间的依赖关系。依赖性评估可通过四项汇总统计来实现。Stoyan的标记-相关,均值-标记,方差-标记和标记-方差图。当分数和分数的分布是独立的时,汇总统计量将保持不变,但如果它们是相关的,则统计量将在不同的距离上变化。通过排列表达值来估计显著性,不同距离间的p值最小即为该基因的显著性。scGCO也利用标记点过程建模空间基因表达,但将HMRF集成到模型中。对于每个基因,scGCO通过图切割算法对图表示进行分割。在完全空间随机框架下,这些片段可以作为候选区域来测试表达式对空间位置的依赖性,其中点在二维空间中的分布被建模为齐次泊松过程。
除标记点法外,还有许多方法采用高斯过程(GP)来模拟空间基因表达。GP是以时间或空间为索引的随机变量集合,其中这些随机变量的有限集合具有多元正态分布。GP在地质统计学中得到了广泛的应用,并应用于空间转录组学建模。例如,SpatialDE基于高斯过程回归,用空间和非空间方差项两部分来模拟每个基因的可变性。可以通过计算这些项的比率来量化空间变异性(Svensson et al, 2018)。通过比较全模型与无空间协方差的零模型的似然,可以用对数似然检验估计统计显著性。SpatialDE可以通过将拟合线性或周期(即余弦)协方差函数的完整模型与高斯核的模型进行比较,进一步识别具有不同类型空间变异的基因,包括线性或周期性模式。为了满足高斯分布的假设,SpatialDE采用了两步归一化。具体来说,SpatialDE使用一种方差稳定变换方法,即Anscombe变换,对nb分布的原始计数进行变换,然后对对数总数进行回归。Gpcounts也建立在高斯过程回归的基础上,但通过NB或零膨胀负二项(ZINB)分布而不是高斯分布来拟合空间计数。同样,BOOST-GP通过ZINB分布来模拟基因读取计数,但采用贝叶斯框架来推断参数。另一种方法SPARK采用广义线性空间模型(GLSM), GP建模空间位置之间的空间关系,泊松分布建模表达式计数数据。
此外,SPARK提供了一种更强大的统计方法来控制I类错误,它分别计算每个参数化核的p值,并将它们与Cauchy组合规则组合在一起。随着ST技术的发展,需要对以往的方法进行改进,以适应高稀疏度的大规模空间转录组学数据。SpatialDE2基于SPARK,通过用omnibus test替代Cauchy combination,并引入Tensorflow的GPU加速来提高计算效率。为了降低计算复杂度和物理内存需求,SPARK的作者提出了一种可扩展的非参数测试方法SPARKX。具体来说,SPARK-X建立在一个非参数协方差测试框架之上,其中计算两个协方差矩阵,分别测量表达相似性和空间接近性。然后将识别具有特定空间趋势的基因转化为检测基因表达与空间位置的相关性。另一种方法是SOMDE,它将自组织映射(SOM)神经网络整合到SpatialDE的高斯过程回归框架中。SOMDE将原始空间位置浓缩为SOM节点,保留空间表达模式和拓扑结构。然后将原始空间表达聚合形成节点级基因元表达,显著减小协方差矩阵的大小,从而提高计算效率。
2)基于图表示
正如在空间域识别一节中所讨论的,空间表达式可以用图表示。一些基于图形的方法已经被证明在SVG识别中是成功的。图拉普拉斯分数通常用于基于图的特征选择,可用于从图中识别空间变量基因。例如,GLISS首先建立一个相互最近邻居图,并计算每个基因的拉普拉斯分数,以测量其位置保持能力(即与局部结构的关联)。在固定的图中,低的拉普拉斯分数表明基因表达的相似性发生在较近的位置,而较大的差异发生在较远的位置。每个基因的统计显著性是通过排列表达和固定的图来估计的。RayleighSelection提出了组合拉普拉斯分数,并将基于图的表示扩展到空间表达数据的简单复杂表示。除了图中包含的顶点和边,简单复合体还包含高维元素,如三角形和四面体,可以捕获更复杂的数据关系。因此,组合拉普拉斯分数有助于识别具有更复杂空间结构的基因。
有些方法在普通的图表示中引入空间网格来简化或优化空间结构。singleCellHaystack是一种基于空间网格的方法,最初用于预测从scRNA-seq学习的低维空间中差异表达的基因,独立于细胞聚类。它还可以应用于使用自然二维或三维空间的空间转录组学数据的SVG识别。singleCellHaystack首先将多维空间划分为网格,并定义网格点,用于估计空间中单元格的参考分布。然后,对于每个基因,singleCellHaystack根据二值化表达将所有细胞分为检测组和未检测组,并分别估计细胞分布。随后计算Kullback-Leibler散度,通过与参考细胞分布的比较来衡量基因的散度,并通过排列检验来评估其显著性。MERINGUE是另一种基于空间网格的方法。它首先使用Voronoi镶嵌构造邻域邻接关系,Voronoi镶嵌也用于构建scGCO中的图表示。与k近邻或k互近邻相比,Voronoi镶嵌可以适应不同的邻域大小和距离,在不同细胞类型和非均匀密度的组织中具有更好的稳定性。然后,MERINGUE计算每个基因的Moran’s I来衡量空间自相关性,它表示空间相邻位置之间的表达相关性。Giotto还提供了一种基于空间网格的方法BinSpect。类似地,BinSpect依靠Voronoi镶嵌来确定邻域关系。BinSpect采用统计富集分析,而不是Moran的I。对于每个基因,BinSpect使用k=2的k-means聚类或简单的秩阈值对表达进行二值化。接下来,计算列联表以反映相邻位置之间的表达式依赖关系。然后采用Fisher精确检验来获得优势比和相应的p值。如果一个基因被发现是重要的,它往往在邻近的位置高度表达。
3)基于其他原则
除了基于统计模型或图形表示的方法之外,还有使用完全不同原理的方法。Sepal提出了一个独特的策略扩散理论,将观察到的基因表达谱视为转录物扩散的结果。在模拟的框架内,sepal假设转录本形成结构化模式比达到均匀随机状态需要更多的时间。因此,推断基因表达模式的结构化程度转化为测量模拟系统中的扩散时间。另一种方法,SPADE侧重于识别与形态特征相关的重要基因。SPADE利用CNN从组织学图像中提取潜在图像特征。然后对高维特征进行主成分分析,总结图像特征的空间分布规律。SPADE使用线性模型来发现与图像模式(即pc)相关的基因,这些基因已被证明具有特定的空间趋势。为了模拟基因表达的空间变异,上述方法只考虑位置之间的相对距离,而忽略了特定方向上的变异。SPATA为用户提供了根据先验知识手动定义轨迹轴的选项。对于每个基因,沿着预定义的空间轴拟合多个函数来模拟空间变化模式,包括线性,对数或梯度上升/下降,单峰或多峰函数。在所有函数中,通过对残差求和的比较,选择最适合的函数来表示基因的动态。在空间可变基因被识别后,一些方法通过聚类进一步确定原型基因模式。SpatialDE通过对聚类质心具有空间先验的扩展高斯混合模型,对具有相似空间表达模式的svg进行聚类。
同样,SPARK实现了一种分层聚类算法,将检测到的变量基因分为不同的类别。MERINGUE不是基于表达构建相似性矩阵,而是通过计算空间相互关联指数来推导相互关联矩阵,这是对每对基因的Moran 's I自相关的改进。这构成了分层聚类的基础。GLISS在潜在结构上拟合一个样条模型,其中每个基因可以用拟合的样条系数表示,具有相似基因模式的基因将共享相似的系数。与基于表达的相似度相比,基于样条系数计算基因-基因相似度可以降低与空间变异无关的相关性。然后,GLISS对系数进行谱聚类,将基因聚类成组。
6. 伪时间轨迹分析
从scRNA-seq或ST数据中,我们只捕获细胞基因表达的快照。通过以上的空间域检测或SVG识别,我们可以分别以离散或连续的方式研究空间上的转录动态。之前在scRNA-seq伪时间分析方面的努力为我们提供了仅从表达数据重建细胞状态轨迹的机会。ST带来的附加空间信息通过引入空间维度扩展了原有的伪时间分析。在ST数据上直接应用单细胞伪时间方法可能导致细胞轨迹随时间连续而在空间上不连续。为了解决这个问题,stLearn通过加入空间信息来调整原始的伪时间算法。stLearn首先利用扩散伪时间(DPT)算法从基因表达中预测伪时间。然后结合基于表达式的伪时间和空间距离的差异计算伪时空距离(PSTD)矩阵,并用一个权值来平衡它们。基于PSTD矩阵,stLearn构建了一个有向图,并应用最小生成树算法来确定分支(即推断细胞轨迹)。与其依赖于仅从基因表达推断的初始伪时间轨迹,还出现了几种从组合表达和空间信息预测细胞轨迹的方法。在空间域识别一节中讨论了SpaceFlow,它提供了一个深度学习框架,可以从ST数据中学习低维嵌入。SpaceFlow生成的嵌入可用于利用DPT算法计算伪时空图(pseudo-Spatiotemporal Map, pSM),便于从ST数据中综合重建时空轨迹。因此,SpaceFlow生成的时空顺序在空间和伪时间上都保持一致性。
7. 细胞-细胞通讯和基因-基因相互作用
通过上述分析,我们可以对细胞类型的空间分布和表达的空间变化有一个基本的了解。然而,细胞或细胞类型的组织,以及产生这种空间模式的基因调控,仍然难以捉摸。许多研究报道,细胞行为可以由来自环境的细胞信号通路塑造。空间转录组学提供了一个独特的机会来研究保存微环境中的细胞-细胞通讯。利用ST数据探索细胞间空间依赖性的方法有几种,其中最直观的方法是研究不同细胞类型的邻近或共定位。例如,Giotto采用随机排列策略来识别富集的细胞型对。在邻域网络结构固定的情况下,对节点间的细胞类型标签进行洗牌,形成随机的邻域关系。通过这种方式,可以确定两种细胞类型之间观察到的超期望频率的比率,并可以估计相应的富集显著性。spicyR最初是为原位细胞术的空间分析而设计的,它定义了一个分数来衡量细胞类型共定位的程度。spicyR通过标记点过程模拟细胞的空间分布,应用k函数或方差稳定的k函数(即l函数)来量化特定距离内两种细胞类型之间的共定位。最近,ang等人基于集体最优运输方法开发了COMMOT,用于处理复杂的分子相互作用和空间约束,以推断空间分辨转录组学中旁分泌依赖的细胞-细胞通信。
除了观察到的细胞类型的共定位外,细胞之间的空间依赖关系可能更为复杂,需要通过更复杂的方法来建模。NCEM在以节点为中心的表达模型中协调方差归因和细胞-细胞通信。NCEM首先使用图结构对单元通信实施邻域约束。通过提供的细胞类型标签,NCEM根据细胞类型和空间背景应用一个功能来拟合细胞所观察到的基因表达。为了适应不同场景中空间依赖关系的复杂性,NCEM提供了三个模型,包括线性、非线性和生成潜变量模型,分别由线性回归、非线性编码器-解码器GNN和条件变分自编码器实现。通过对目标细胞(即接收器)的分子状态对邻域(即发送方)的依赖性进行建模,NCEM还可以确定发送方-接收器信号的方向性。不同于对整个表达谱依赖于细胞间通讯的建模,有几种方法量化了细胞间相互作用对每个基因表达的影响。例如,SVCA使用高斯过程模型对靶基因在细胞间的表达进行建模,并将基因的变异性分解为三个组成部分,包括内在效应、来自未测量空间变量的环境效应以及来自邻近细胞的细胞间相互作用效应。通过这种方式,可以估计每个基因的每个术语解释的方差的比例,并且可以识别参与细胞-细胞相互作用的生物学相关基因。MISTy设计了一个多视图框架来解释单个基因的表达,其中来自不同空间背景的细胞-细胞相互作用在不同的视图中建模。与SVCA相似,MISTy包括内观、近观和旁观,分别对应于同一位置其他基因表达的内在影响、近邻的影响和组织结构(即指定细胞半径内的细胞)的影响。通过分析每个预测基因在每个视图中对目标基因的重要性,不同空间背景的影响可以解释感兴趣的基因对。SVCA和MISTy可以模拟基因-基因关系,发现与细胞-细胞相互作用相关的基因,但它们都不能识别显性基因-基因相互作用对。Yuan和Bar-Joseph开发了GCNG,一种基于gcns的监督计算框架,用于预测基因相互作用。GCNG以空间邻域的图表示作为输入,以及候选基因对的归一化表达。输出将是相互作用或非相互作用基因对的分类。
为了实现监督学习,已知的配体-受体相互作用被标记为正对,随机选择的配体-受体对被标记为负数据。GCNG具有五层GCN结构,可以预测所研究的ST数据集中新的基因-基因相互作用。然而,GCNG不能告知相互作用发生的细胞类型,也不能关注特定局部区域内的相互作用推断。为了解决这些局限性,一些方法通过考虑细胞类型位置依赖于配体和受体的共表达。例如,MERINGUE进一步将基因对之间的空间互相关计算限制为筛选的配体-受体对和两种感兴趣的细胞类型。Garcia-Alonso等人将他们的CellphoneDB升级到v3.0,可以在特定的微环境中识别感兴趣的细胞类型的配体-受体对。同样,在上一步细胞类型接近分析的基础上,Giotto通过计算相互作用细胞类型的细胞亚群中配体和受体的加权平均表达来定义配体-受体相互作用评分。
8. 空间数据综合分析
随着通量的增加和成本的降低,一些研究从多个个体中生成ST幻灯片以进行大规模分析。其他一些研究从组织的多个相邻层产生一系列ST玻片,从而实现整个组织的全局视图。对单个ST玻片进行单独分析可能会降低多个样品的功效。因此,需要采用积分方法对多个样本进行联合分析。此外,随着形态学等附加信息的提供,空间转录组学应该与其他模式相结合,以全面表征组织。在本节中,我们将回顾多样本集成和不同模态空间数据集成的计算方法。
1)多试样集成
多样本积分的核心是将多个样本放置在同一空间,称为共同坐标框架(common coordinate framework, CCF)。坐标系包含两个方面。一方面,CCF可以代表自然的三维空间,其中多个平面幻灯片排列堆叠,提供组织的立体视图。另一方面,来自多个样本的高维空间位置测量可以投影到共享的低维空间中,用于联合空间域识别等综合分析。
已经开发了一些方法来排列来自同一组织的多个连续载玻片。PASTE将多片排列表述为一个最优运输问题,该问题基于基因表达和空间信息计算概率排列。通过最小化运输成本,PASTE可以实现最大限度地提高幻灯片上对齐位置之间的基因表达相似性,同时保留幻灯片内的空间结构。PASTE可以对齐来自同一组织的多个连续幻灯片,但不能应用于来自不同时间点的幻灯片的集成。Andersson等人提出了一种方法eggplant,这是一种基于地标的方法,将多个幻灯片投影到共同参考文献中。首先,eggplant将测量到的空间位置投影到参考点上,使变换前后地标之间的距离保持不变。接下来,eggplant应用高斯过程回归来学习所有排除地标的位置的基因表达与到地标的距离之间的关系,从而可以预测参考中每个位置的基因表达。采用位置转换与表达预测相结合的策略,可以将不同时间点或不同个体的多张幻灯片转移到同一参考文献中进行综合分析。然而,eggplant不仅需要选择地标位置,还需要定义参考,参考通常是代表组织域的规范结构。这两个要求限制了eggplant在肿瘤等更复杂组织上的应用。为了解决这个问题,Jones等人开发了GPSA,它也是基于高斯过程模型。GPSA构建了一个双层高斯过程框架,其中第一层将测量的空间位置映射到一个公共坐标系,第二层描述该系统内的空间基因表达。与eggplant相比,GPSA可以从头迭代估计公共坐标系统,但它也提供了基于模板的与预定义的公共坐标对齐的选项系统通过固定一个幻灯片。
不同于将空间位置从多张幻灯片映射到自然3D空间中的CCF,有几种方法侧重于将多个样本投影到共享的低维空间。在这种情况下,整合方法应该能够从不同批次中去除不需要的变异,并保留有意义的生物变异,如scRNA-seq。但不同于单细胞积分法,ST积分法需要考虑空间信息。Liu等人提出了PRECAST,这是一种统一的原则性概率模型,用于联合估计低维嵌入并在多个组织载片上执行空间聚类。PRECAST采用内禀条件自回归(CAR)模型对归一化的基因表达进行降维,在低维空间中保持了邻居之间原有的空间依赖性。由此产生的潜在低维嵌入可以进一步利用HMRF模型进行空间聚类。正如我们上面提到的,BASS支持多尺度分析,同时进行细胞类型聚类和空间域检测。它还允许多样本整合分析,通过联合建模和谐校正的空间转录组与层次贝叶斯框架。另一种方法,MAPLE提出了一个混合框架,用于多个部分的联合空间聚类,通过基于gcn的模型进行空间感知的低维嵌入学习。
2)多模态集成
如上所述,单细胞和空间转录组学通常结合起来,通过细胞作图或细胞类型反褶积来破译细胞类型的空间分布。在我们回顾的整合方法中,Tangram脱颖而出,通过与多模态单细胞数据集成,将其他模式的数据映射到空间转录组学。例如,一旦SHAREseq的单细胞通过基因表达相似性定位到空间位置,染色质可及性的空间模式就可以被揭示。考虑到许多ST技术提供相应的组织学图像,许多计算方法利用额外的图像信息来提高每一步的分析性能。例如,stLearn利用形态相似性对表达数据进行规范化,从而减少技术噪声的影响。在计算点-点距离构建空间转录组学图时,spaGCN考虑了组织学图像特征。stMVC采用带有注意机制的图网络来整合包括组织学特征在内的多源信息,最终学习ST数据的低维嵌入。同样,conST和MUSE等方法也使用深度学习架构来整合细胞形态和转录状态以进行联合表示。SPADE没有采用复杂的基于深度学习的机制,而是使用线性回归模型直接将基因表达的空间方差与图像特征的空间分布模式联系起来。
除了便于空间转录组学分析外,组织学图像还可用于预测空间基因表达。已经开发了许多方法来解决这个问题。为了克服一些基于条形码的ST技术的低分辨率限制,bergenstratuhle等人提出了一种深度生成模型,从高分辨率组织学图像中推断超分辨率表达图,包括原始测量位置内部和之间。一些方法不是专注于提高空间基因表达的分辨率,而是将空间转录组预测推广到没有匹配表达数据的组织病理图像。例如,He等人引入了一种深度学习算法ST-Net,通过结合空间转录组学和组织学图像来捕获基因表达异质性。该模型使用包含68个乳腺组织切片的ST切片的BRCA空间转录组数据集进行训练,可以直接从组织学图像中预测其他乳腺癌数据集的空间分辨率转录组。然而,ST-Net并没有考虑到点之间的空间依赖性。HisToGene采用了一种改进的Vision Transformer模型来预测空间基因表达,并考虑了位点依赖性。在HisToGene的基础上,Hist2ST还包括一个Convmixer模块,用于捕获图像斑块内2D视觉特征的内部关系。
应用程序
近年来空间转录组学的快速发展促进了其在各种生物系统中的广泛应用。ST技术在空间表征健康组织的细胞状态方面发挥了重要作用,其中一些技术旨在破译特定发育阶段组织的空间结构。值得注意的是,在这些组织中,神经系统一直是研究的焦点。许多研究为构建详细的大脑空间图谱做出了重大贡献。此外,ST技术在探索损伤或病变组织的微环境方面已被证明是无价的,包括感染病毒的小鼠肺部,心肌梗死的人类心脏以及一系列不同的肿瘤类型。本文综述了ST在三个主要领域的应用,包括健康组织的发育和动态平衡、神经科学和肿瘤微环境。
1)健康组织的发育和体内平衡
大多数研究利用小鼠模型来研究早期哺乳动物胚胎的发育。已经建立了小鼠胚胎发育的几个阶段的空间图谱。Peng等人关注着床后阶段的谱系分化和形态发生。Geo-seq应用于从原肠形成前(胚胎期第I5.5天)到原肠形成后期(胚胎期第7.5天)所有胚层中预先选择位置的细胞群。该研究揭示了谱系规范和组织模式在时间和空间上的动态分子调控。此外,他们还发现了Hippo/Yap信号在细菌层发育过程中的关键作用。为了进一步探索原肠胚形成结束时早期器官发生中细胞命运的决定,Lohoff等对E8.5 - E8.75时收集的小鼠胚胎的多个矢状切片进行了seqFISH。由于目标基因数量有限,他们将seqFISH与现有的单细胞转录组图谱整合,以实现全基因组的植入。利用生成的单细胞空间图谱,揭示了早期发现的中脑和后脑区域背-腹轴和喙-尾轴对应的基因表达空间模式肠管背腹分离。最近,Chen等人将高分辨率Stereo-seq技术应用于E9.5 ~ E16.5妊娠中后期的全鼠胚胎,最终构建了小鼠器官发生时空转录组图谱(MOSTA)。除了小鼠的早期胚胎发育,许多研究人员已经利用空间转录组学来探索驱动人类器官或组织发育的空间依赖机制。例如,Crosse等人利用基于lcm的RNA测序技术,对卡内基阶段(CS)16 - CS17(即受孕后39-41天)的人类胚胎中正在发育的造血干细胞(HSC)生态位进行了空间分辨分析。他们分析了主动脉的背腹侧极化信号,并确定腹侧分泌的内皮素是早期人类HSC发育的重要分泌调节因子。在对人类心脏发育的研究中,Asp等人使用ST技术表征了人类心脏在三个发育阶段(受孕后4.5 - 5周、6.5周和9周)的不同解剖区域。通过scRNA-seq和ISS的整合,建立了一个全面的空间图谱,提供了人类心脏发生过程中细胞亚型定位的详细信息。类似的策略也应用于8至22 PCW的人类肠道发育研究。除了生成人类肠道发育的时空图谱外,他们还揭示了形态梯度如何指导细胞分化。空间转录组学也被应用于成人健康组织的细胞型图谱和稳态维持的研究,可以作为与病变组织比较的参考。Shen等利用Stereo-seq技术绘制了人类牙龈的ST图谱。通过鉴定牙周炎相关效应细胞、基因和途径,ST结果可能有助于开发新的牙周炎治疗策略。Madissoon等人通过结合scRNA-seq、snRNA-seq和10x Visium ST,创建了人类肺和气道的空间多组学图谱,其中包括各种新的和已知的细胞类型。空间肺图谱还揭示了特定的组织微环境,如腺体相关淋巴细胞生态位(GALN),可能在预防呼吸道感染中发挥作用。在另一项关于人类子宫的研究中,Garcia-Alonso等也应用多组学技术构建了人类子宫内膜的综合细胞图谱,表征了整个月经周期的时空动态。特别是,进一步的空间相互作用分析揭示了NOTCH和WNT信号通路在塑造纤毛和分泌细胞系分化中的作用。随着ST数据的积累,可以预见,在不久的将来,多源组织图的整合将导致整个人体综合参考空间图谱的建立。
2)神经科学
明确的分层结构和独特的解剖区域使大脑成为验证新开发的空间转录组学技术的合适材料。反过来,这些ST技术显著增强了我们对大脑空间结构的理解。许多研究都致力于构建大脑的参考图。由于早期基于成像的ST技术的视野大小有限和密集劳动性质,大多数研究集中在小鼠大脑的特定亚区域。例如,Codeluppi等人开发了osmFISH,并使用该方法定义了体感觉皮层的空间细胞组织,仅涵盖33个靶向标记基因和约5000个细胞。与此同时,Moffitt等人通过将MERFISH与scRNA-seq相结合,生成了下丘脑视前区神经元的空间分子图谱。同样,大脑的其他亚区,如视觉皮层、初级运动皮层、海马和小脑,已经通过不同的基于成像的ST技术来建立详细的空间细胞组织图。由于基于高通量条形码的ST技术的发展,Ortiz等人建立了整个成年小鼠大脑的分子图谱。他们利用ST技术分析了从一个大脑半球沿前后轴收集的75个邻近冠状面切片的空间基因表达。通过与Allen小鼠脑图谱(ABA)的比对,他们构建了一个完整的脑图谱,提供了三维组织坐标和详细的ABA神经解剖学定义。更重要的是,他们还通过无监督分类在分子图谱中定义了新的区域和层特异性亚区。无论是整个大脑还是特定的子区域,这些图谱对实验神经科学都有很大的价值,最终扩展了我们对大脑结构-功能关系的认识。
除了揭示正常大脑中细胞类型的空间组织外,空间转录组学还可以扩展到神经退行性疾病或精神疾病的研究,揭示神经系统功能障碍或失调的空间相关机制。例如,Chen等将ST技术与ISS相结合,捕捉了阿尔茨海默病(AD)淀粉样斑块附近的转录变化。特别是,他们确定了两个基因共表达网络,可能对阿尔茨海默病中的淀粉样斑块沉积有反应。Maniatis等在肌萎缩性侧索硬化症(ALS)的研究中,利用不同阶段的ALS小鼠模型,采用ST技术表征疾病进展的时空动态。结合ALS患者的死后组织,他们在与ALS病理相关的转录途径中发现了共同的空间扰动模式。随着ST技术在分辨率和检测效率方面的不断提高,我们期望建立更详细和全面的神经系统地图集。这些资源对于探索电路和行为的结构-功能关系无疑是无价的。
3)肿瘤微环境
尽管单细胞转录组学已经揭示了复杂的TME中的细胞类型组成及其功能,但仍未探索这些细胞如何在空间上组织以控制或促进肿瘤进展。空间转录组学使研究不同的细胞群体和信号通路与空间背景保存成为可能。肿瘤微环境一般包括肿瘤细胞、基质细胞和免疫细胞。最初的研究工作往往集中在肿瘤区域的内部异质性。在人类乳腺癌(BRCA)的单细胞和空间图谱研究中,Wu等人从scRNA-seq中衍生出7个基因模块来描述肿瘤内的转录异质性。分析发现两个在肿瘤区域相互排斥的基因模块,它们分别与EMT和增殖状态有关。在另一项原发性肝癌的研究中,定义了5种癌症干细胞(cancer stem cell,CSC)群体,它们在不同的区域表现出不同的分布模式,包括前沿、肿瘤和高级别门静脉肿瘤血栓形成。值得注意的是,PROM1+ CSCs在门静脉肿瘤血栓中的比例高于肿瘤区域,可能在肿瘤进展中发挥关键作用。
以肿瘤区域为中心,利用空间转录组学可以揭示免疫或基质细胞类型的相对空间分布。Ji等在人鳞状细胞癌(SCC)的研究中发现,B细胞在肿瘤中浸润,而在肿瘤-间质边界大量存在调节性T细胞、巨噬细胞和成纤维细胞。相反,CD8 T细胞明显被排除在肿瘤之外。同样,不同的细胞亚型或状态也显示出不同的空间模式。Wu等在乳腺癌的TME中发现了炎症样癌症相关成纤维细胞(iCAFs)和肌成纤维细胞样CAFs (mCAFs),但这两种亚型表现出明显不同的空间分布。mCAFs富集于浸润性癌区,而iCAFs分散分布于浸润性癌区、间质区和淋巴细胞聚集区。一些研究感兴趣的是肿瘤-基质边界的分子和细胞类型模式,即肿瘤的侵袭。Wu等人描述了侵袭中细胞类型丰度的动态变化,并在边界附近区域发现了免疫抑制微环境。
空间分析还可以识别肿瘤微环境中的一些模式结构并对其进行表征。在上述肝癌研究中,对ST点进行无监督聚类,发现了一个以三级淋巴结构(TLS)相关基因高表达为特征的聚类,如CXCL13、CCL19、CCL21、LTF和LTB。病理检查证实存在TLSs。Wu等定义了一种TLS-50特征来定位其他组织切片中的tls,也发现TCGA的HCC患者预后较好。同样,Andersson等也在her2阳性乳腺癌中发现了TLSs。为了进一步研究TLS如何影响癌症对免疫治疗的反应,梅兰等人使用空间转录组学研究了肾细胞癌(RCC)中TLS内B细胞反应的性质。他们发现TLSs可以产生和繁殖产生抗肿瘤抗体的浆细胞,这与免疫治疗的反应有关。已知细胞通讯在肿瘤的免疫监视或逃逸以及肿瘤进展中发挥重要作用。通过细胞类型反褶积或细胞作图分析揭示细胞类型的空间分布,还可以识别细胞类型接近或共定位模式。
Moncada等人通过将scrna -seq定义的细胞类型定位到胰腺导管腺癌的ST,确定了炎症成纤维细胞和应激反应癌细胞的共定位。同样,scRNA-seq和ST在SCC中的整合,在肿瘤特异性角化细胞群周围发现了纤维血管生态位。进一步的相互作用分析表明,共定位可能由多个配体-受体对介导。在结直肠癌的另一项研究中,空间转录组学和免疫荧光染色显示FAP+成纤维细胞和SPP1+巨噬细胞共存,这与患者生存率低有关。
随着空间多组学技术的发展,细胞串扰和代谢状态等其他方面的特征将被表征,以获得对肿瘤微环境复杂性的更多见解。了解肿瘤微环境有助于确定治疗靶点和设计抗肿瘤药物。
总结
本章全面概述了当前空间转录组学的进展,包括技术创新、计算方法和各种应用。空间转录组学彻底改变了我们对组织组织和细胞异质性的理解,使完整组织内基因表达模式的高分辨率可视化成为可能。计算方法的发展促进了空间转录组学数据的整合和解释,揭示了空间调控机制和新的分子相互作用。空间转录组学已经成功地应用于各个领域,包括发育生物学、神经科学、癌症研究和免疫学,具有加速生物标志物发现和个性化医学方法的潜力。空间转录组学代表了一种变革性的方法,并将继续完善以重塑我们对复杂生物系统的理解。我们预计它将为组织稳态和疾病机制提供深刻的见解。
参考文献:
Sun F, Li H, Sun D, et al. Single-cell omics: experimental workflow, data analyses and applications. Sci China Life Sci. 2025;68(1):5-102. doi:10.1007/s11427-023-2561-0